MuSpAn with Xenium Explorer#
In this tutorial, we will introduce some handy helper functions that have been developed to import spatial transcriptomics data generated using the Xenium platform. These functions are designed to improve the workflow of spatially analysing these very large and multiscale Xenium datasets, and specifically to be used in tandem with the 10x Xenium Explorer software to focus on regions for analysis.
To get started, we will need a Xenium dataset to import and analyse. I will be using a sample dataset from 10x of Healthy Mouse Colon, produced using the in situ gene expression with cell segmentation staining data for mouse colon tissue using the Xenium mouse tissue atlassing panel, which can be found on the 10x resources site.
The complete dataset folder is saved to my local machine, which is important as we will need all output files (cells.parquet,cell_boundaries.parquet, transcripts.parquet,nucleus.parquet, experiment.xenium) to unpack the spatial information to create a MuSpAn domian.
As there is so much information in these datasets, we will use Xenium Explorer to annotate regions of interest as shown below:

We downloaded the Cell Stats as CSV from the Selections tab in Xenium Explorer, saving it under the filename ‘mouse_region_for_tutorial.csv’ in a local directory. Make sure you know where is has been saved as we will need to use this file to create a MuSpAn domain.
Note: The cell clusters willl be defined by the clustering method that is currently actively in Xenium Explorer as we retreive the cluster information from the region-of-interest csv file (mouse_region_for_tutorial.csv). In this example, we have the ‘graph-based’ cell clustering selected which will imported as labels for the cells.
Now We have everything we need to create a MuSpAn domain! So let’s set up our usual imports
[2]:
# Import necessary modules
import muspan as ms # MuSpAn for spatial analysis
import numpy as np # NumPy for numerical operations
import matplotlib.pyplot as plt # Matplotlib for plotting
Functions for tranforming data into MuSpAn domains are contained in the muspan.io module and so we can import our Xenium data using the ms.io.xenium_to_domain function. We have a few option when importing the data on the format of the objects (do we want cells as points or shape) and the option to import nuclei segmentations. Importing transcripts needs slightly more care as there can be a lot of them. Therefore if you are loading in transcript data, we suggest you specify transcript names
of interest to not flood the memory of your machine.
Here we are going to load in the cells as shapes, with their nuclei and selection of transcripts the we may be interested in:
[ ]:
# Define the path to the Xenium data and the selection region CSV file
path_to_my_xenium_data = 'path/to/Data/Xenium_V1_mouse_Colon_FF_outs' # Path to the Xenium data
path_to_my_selection_region = 'path/to/Data/mouse_region_for_tutorial.csv' # Path to the selection region CSV file
# List of transcript names of interest
transcripts_of_interest = ['Oit1', 'Cldn2', 'Nupr1', 'Sox9', 'Ccl9', 'Mylk', 'Myl9', 'Cnn1', 'Mgll', 'Mustn1']
# Import the Xenium data into a MuSpAn domain
pc = ms.io.xenium_to_domain(
path_to_xenium_data=path_to_my_xenium_data,
domain_name='A test domain',
cells_from_selection_csv=path_to_my_selection_region,
load_transcripts=True,
selected_transcripts=transcripts_of_interest,
load_nuclei=True,
load_cells_as_shapes=True,
exclude_no_nuclei_cells=True
)
Depending on the resize of the ROI and number of transcripts being loaded, this can take sometime (<5mins). For larger regions, we recommend loading in cells in as points, without nuclei and transcripts. Let check everything has been loaded in correctly:
[9]:
# Print the MuSpAn domain object to check its contents and ensure it has been loaded correctly
print(pc)
Domain name: A test domain
Number of objects: 19693
Collections: ['Cell boundaries', 'Nucleus boundaries', 'Transcripts']
Labels: ['Cell ID', 'Transcript Counts', 'Cell Area', 'Cluster ID', 'Nucleus Area', 'Transcript', 'Transcript ID']
Networks: []
Distance matrices: []
Under the hood, muspan is linking all the data using the Cell ID label. This label is added all to objects loaded in and can be used to query data. In addition, the cell boundaries (or centroids if loaded as points), nuclei and transcripts are allocated as separate collections of objects which can also be queries for analysis. For more information on querying, check out our querying tutorials!
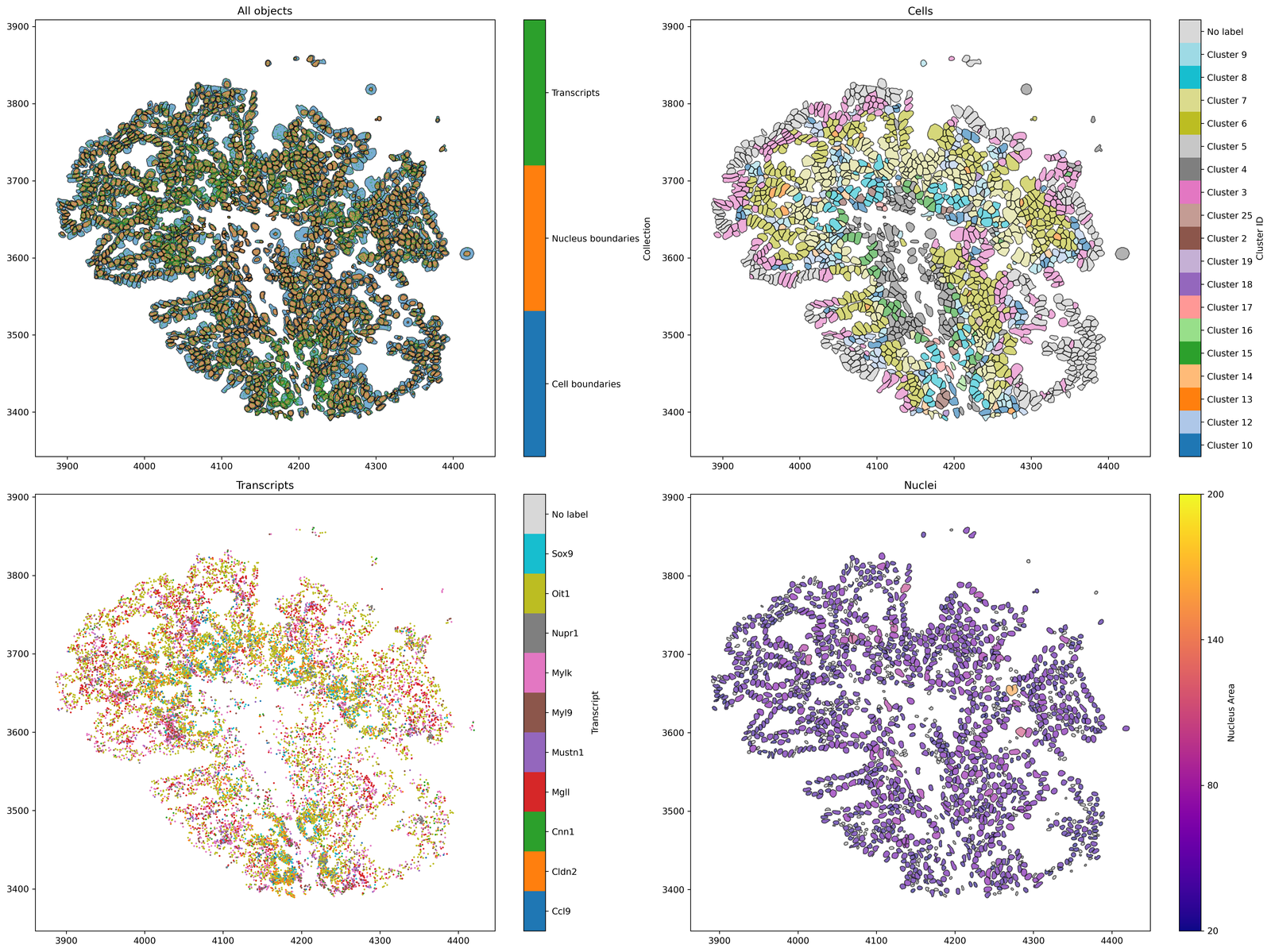
Now we have our domain loaded, let’s visualise the data to make sure it’s what we expected from the Xenium Explorer.
[8]:
# Queries to isolate the different objects within the MuSpAn domain
# Query to isolate cell boundaries
qCells = ms.query.query(pc, ('Collection',), 'is', 'Cell boundaries')
# Query to isolate transcripts
qTrans = ms.query.query(pc, ('Collection',), 'is', 'Transcripts')
# Query to isolate nucleus boundaries
qNuc = ms.query.query(pc, ('Collection',), 'is', 'Nucleus boundaries')
[12]:
# Create a figure with 2x2 subplots
fig, ax = plt.subplots(figsize=(20, 15), nrows=2, ncols=2)
# Visualise all objects in the MuSpAn domain
ms.visualise.visualise(pc, ax=ax[0, 0], marker_size=0.05)
ax[0, 0].set_title('All objects')
# Visualise cells, colored by 'Cluster ID'
ms.visualise.visualise(pc, color_by=('label', 'Cluster ID'), ax=ax[0, 1], objects_to_plot=qCells)
ax[0, 1].set_title('Cells')
# Visualise transcripts, colored by 'Transcript'
ms.visualise.visualise(pc, color_by=('label', 'Transcript'), ax=ax[1, 0], objects_to_plot=qTrans, marker_size=1)
ax[1, 0].set_title('Transcripts')
# Visualise nuclei, colored by 'Nucleus Area'
ms.visualise.visualise(pc, color_by=('label', 'Nucleus Area'), ax=ax[1, 1], objects_to_plot=qNuc, vmin=20, vmax=200)
ax[1, 1].set_title('Nuclei')
[12]:
Text(0.5, 1.0, 'Nuclei')
To allow for multiscale anlysis, we automatically construct object inheritance using the Cell ID. For example, Cells are parents of Transcripts which we can use for linking the spatial scales. This can be viewed using the ms.visualise.visualise_family_tree function.
[18]:
# Define the cell ID of interest (found using Xenium Explorer or by looking at the cell ID in the cell table)
cell_id_of_interest = 'bnbekepb-1'
# Create a query to isolate the candidate cell based on the cell ID
candiate_cell_query_1 = ms.query.query(pc, ('label', 'Cell ID'), 'is', cell_id_of_interest)
# Create a query container and add the candidate cell query to it
query_this_cell = ms.query.query_container()
query_this_cell.add_query(candiate_cell_query_1, 'AND', qCells)
# Get the family tree of the selected cell to retrieve its ancestors
this_cells_acenstors = list(ms.query.get_family_tree(pc, query_this_cell).nodes())
# Create a figure with 1x2 subplots, with different width ratios
fig, ax = plt.subplots(figsize=(25, 4), nrows=1, ncols=2, width_ratios=[3, 1])
# Visualize the family tree of the selected cell, coloring by 'Transcript'
# We can color by any label or collection, see documentation for more details
ms.visualise.visualise_family_tree(pc, query=query_this_cell, ax=ax[0], color_by='Transcript', add_id_label=False, add_cbar=False)
# Visualize the selected cell and its ancestors, coloring by 'Transcript'
ms.visualise.visualise(pc, objects_to_plot=this_cells_acenstors, ax=ax[1], color_by=('label', 'Transcript'))
[18]:
(<Figure size 6750x1080 with 3 Axes>, <Axes: >)

Now we are ready to start analysing the data!